
RESEARCH ARTICLE
Gene Expression of GSK3 in Type II Diabetics Compared to Non-Diabetics (ex vivo)
Somayeh A.H. Khorami1, Mohd S. Abd Mutalib1, Mohammad F. Shiraz2, Joseph A. Abdullah3, Zulida Rejali4, Razana M. Ali5, Huzwah Khaza’ai6, *
Article Information

Identifiers and Pagination:
Year: 2020Volume: 10
First Page: 30
Last Page: 37
Publisher Id: TODIAJ-10-30
DOI: 10.2174/1876524602010010030
Article History:
Received Date: 06/05/2020Revision Received Date: 21/10/2020
Acceptance Date: 26/10/2020
Electronic publication date: 23/12/2020
Collection year: 2020
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background:
GSK3 is a serine/threonine kinase that is involved in the storage of glucose into glycogen through the negative regulation of glycogen synthase. Defects in GSK3 and glycogen synthase function are early stages of the development of insulin resistance, which may cause impaired glycogen synthesis in Type II diabetes.
Methods:
In this cross-sectional study, the gene expression level of GSK3 from Type II diabetic and non-diabetic participants was compared via real-time RT-PCR. To investigate the relationships between GSK3 expression and indicators of insulin resistance, Pearson's correlation analysis was performed. To compare the differences between GSK3 expression levels based on BMI categories, one-way ANOVA was used.
Results:
Gene expression of GSK3 was slightly higher in diabetic participants compared to non-diabetics, but it was statistically insignificant. Also, no significant difference was found based on BMI categories in the two groups. No significant association between GSK3 expression and indicators of insulin resistance was observed in non-diabetic participants. There was only a positive significant correlation between GSK3 expression and FBS in diabetic participants.
Conclusion:
These results indicate that the regulation of GSK3 may occur at the translation level, as gene expression level was unaltered between diabetic and non-diabetic participants. Also, since circulating levels of both glucose and insulin regulate GSK3 activity, tissue specificity for the expression and post-translation regulations of GSK3 may exist, which cause hyperactivation or overexpression in some target tissues in diabetes. Furthermore, it is probable that glycogen synthase activity is also regulated by non-insulin mediated mechanisms like exercise or allosteric changes, independent of GSK3 expression.
1. INTRODUCTION
Impaired insulin regulation of glucose transport and utilization is associated with insulin-resistance and diabetes [1]. Insulin-induced glycogen synthesis in elevated postprandial blood glucose levels is necessary to maintain glucose homeostasis and defective regulation of glycogen synthesis by insulin is an additional feature of the insulin-resistant condition [2]. Insulin receptors in target tissues stimulate a signaling cascade leading to phosphorylation/activation of insulin receptor substrates (IRS1 and IRS2) and subsequent activation of phosphatidylinositol-3 kinase (PI3K) cascade, which is the main pathway involved in glucose transport and glycogen synthesis [3]. By this signaling axis, insulin stimulates phosphorylation of phosphoinositide-dependent serine-threonine protein kinase (AKT) and Glycogen synthase kinase-3 (GSK3), which increases and decreases their activities, respectively [4]. The PI3K/AKT pathway is antagonized by various factors, including phosphatase and tensin homolog (PTEN) [5]. Glycogen synthase (GS) activity is activated by PI3K/AKT signaling cascade and inhibited by GSK3 through phosphorylation. AKT phosphorylates/inactivates GSK3 and increases GS activity [3]. GSK3 is a serine/threonine-protein kinase and a negative regulator of glycogen synthesis and lipogenesis [6]. Previous in vitro studies have demonstrated the role of GSK3 in the regulation of glycogen synthase phosphorylation and its function [7]. A specificity of GSK3 regulation is that it is constitutively activated [8]; thus, the negative regulation of GSK3 through the PI3K/AKT pathway keeps GSK3 activity at low stages [9]. Several disease conditions, such as insulin resistance, can break cellular homeostasis and lead to increased GSK3 activity [10]. Overly activated GSK3 in disease conditions may occur when the PI3K/AKT pathway is either over-stimulated (e.g., chronic over-nutrition) or repressed as a result of inhibitors or lack of stimuli. This “overstimulation-induced insensitivity” phenomenon is commonly present in almost all of the metabolic disorders [11]. Serine phosphorylation of GSK3 by AKT results in inhibition of its kinase activity, whereas tyrosine phosphorylation of GSK3 promotes the activity of the enzyme [11]. Also, GSK3 phosphorylates IRS on serine residues and causes its proteolytic breakdown [12], which is one of the negative-feedback loops implicated in the PI3K/AKT signaling pathway that down-regulates insulin signal transduction. Furthermore, GSK3 plays the main role in PTEN phosphorylation, which acts as an antagonist factor of insulin signal transduction [13]. In addition, it has been demonstrated that GSK3 is suppressed by physiological concentrations of insulin in human skeletal muscle [14, 15] as well as different cell lines [16-18]. As GSK3 is inhibited by insulin (to stimulate the PI3K/AKT signalling pathway), phosphorylation and deactivation of PTEN by GSK3 might be part of a negative feedback loop for the PI3K/AKT pathway [4] (Fig. 1).
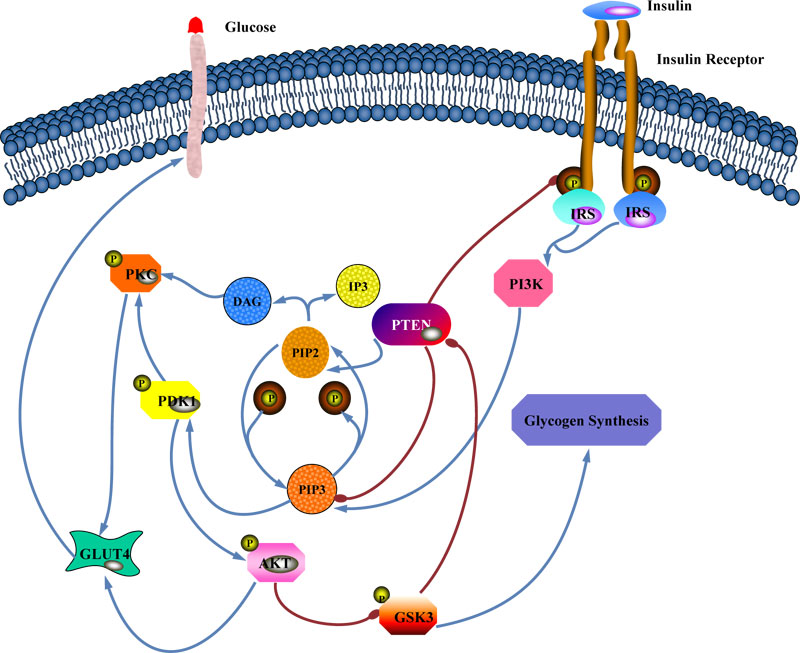 |
Fig. (1). Proposed negative feedback loop to regulate the PI3K/AKT pathway. |
Administration of GSK3 inhibitor increased glucose tolerance in diabetic mice [19, 20] and reduced blood glucose levels in patients with Type II diabetes as well as in Zucker diabetic fatty rats [21-24]. These findings demonstrate that inhibition of GSK3 improves glucose regulation in diabetic animals. Consistent with this, overexpression of GSK3 is enough to cause glucose intolerance [19, 25].
Since it has been suggested that GSK3 is a ubiquitous kinase implicated in insulin function that induces insulin resistance and diabetes through different pathways, it was considered in the present study. The aim of this study was to investigate the relationships between GSK3 gene expression levels and indicators of insulin resistance and compare those between diabetic and non-diabetic individuals.
In this schematic, insulin activates the insulin receptor and PI3K. The resulting increase in PIP3 level activates the AKT kinase that phosphorylates and inhibits GSK3. The loss of GSK3 activity enhances PTEN activity, resulting in a negative feedback loop.
2. MATERIALS AND METHODS
2.1. Experimental Participants
The present work is a cross-sectional study that included 50 Type II diabetic and 50 non-diabetic individuals in the age group of 35-60 recruited from University Putra Malaysia and Serdang Hospital. The number of men and women were equal in each group and there was no statistically significant difference between the two groups with respect to age (P>0.05). Diabetic and non-diabetic participants were divided into two groups on the basis of body mass index (BMI<30 and BMI>30). In diabetic and non-diabetic groups, 50% were normal/overweight (BMI <30 kg/m2) and 50% were obese (BMI of >30 kg/m2). This weight distribution was not significantly different between the groups. All diabetic patients were administered to the anti-diabetic drug Metformin without insulin injection. Participants who had cancers, diabetic patients with diabetes complications, and pregnant women were excluded from this study. Ethical approval was obtained from the University Putra Malaysia Research Ethics Committee and the National Medical Research Register (Ministry of Health). The study was monitored according to the Declaration of Helsinki in its currently applicable version and the guidelines of the International Conference on Harmonization of Good Clinical Practice. Informed written consent was obtained from all participants.
2.2. Assessment of Clinical and Metabolic Characteristics of Subjects
FBS (Cat.No:04657527) was tested by the colorimetric enzymatic method using Roche Diagnostics GmbH Cobas-c 311 Germany machine. HbA1c analysis was performed by the colorimetric method (Diazyme; cat. No. DZ168A, USA) and the C-peptide level was determined by ELISA Kit (Cloud Clone Corp; cat. no. CEA447Hu, UK).
2.3. RNA Extraction and Real-time PCR
Real-time RT-PCR method was employed to investigate the variability within non-diabetic and Type II diabetic participants in gene expression of GSK3 with a set of two housekeeping genes (GAPDH and β-ACTIN). The analysis was performed using the three technical replicate samples from each of the participants. Briefly, RNA was extracted from 2.5 ml blood by using PAXgene Blood RNA Tubes (Qiagen; cat. no. 762165). RNA concentration and purity were measured using a NanoDrop ND1000 spectrophotometer at A260: A280 ratio (Thermo Fisher Scientific; USA). RNA quality and integrity were determined via agarose gel electrophoresis. The purified RNA sample was converted to cDNA, using the Qiagen cDNA Synthesis Kit, according to the manufacturer’s instructions (Qiagen; cat. no. 205313), and then cDNA was used to perform RT-PCR via SYBR green technology with the QuantiTect Reverse Transcription Kit (Qiagen; cat. no. 204054). The final volume of the RT-PCR reaction mixture was 20 µl and contained 2 µl cDNA, 1 µM of each primer, 10 µl Master Mix, and RNase-free water up to 20 µl. GAPDH and β-ACTIN were chosen as suitable reference genes to normalize the mRNA expression. The specificity of the product was assessed from the melting curve analysis. According to manufacturer recommendation, real-time PCR quantification experiments should include a positive control containing all the components of the reaction except for the template (NTC) and negative control (template without reverse transcriptase) to ensure the quality of run and confirm the absence of contamination. In this study, primers were designed by Qiagen and cycling conditions were as follows: 95 ºC for 5 min (PCR initial activation step); 95 ºC for 10 s (denaturation); 60 ºC for 30 s (combined annealing/extension); followed by 40 cycles and it was performed on a Bio-Rad Real-Time PCR detection system (Bio-Rad; CFX96, USA). Average gene Ct values were normalized to the average of housekeeping genes Ct values of the same cDNA sample. Fold differences were determined using the comparative ∆∆CT method.
2.4. Statistical Analyses
Data are shown as means ± SE unless stated otherwise. Before statistical analysis, non-normally distributed parameters were logarithmically transformed to approximate a normal distribution. Statistical analysis was performed using independent-samples t-test with the SPSS 21.0 statistical software package (SPSS Inc., Chicago, IL, USA). The threshold of significance was defined as a P<0.05. Pearson's correlation was used as appropriate to analyze the relationship between measured biochemical markers and expression level of GSK3. One-way ANOVA was used to compare differences of GSK3 expression between two groups based on BMI categories. Significance was accepted at a P<0.05. Levene's test was used to check significant differences revealed by the ANOVA.
3. RESULTS
The clinical and metabolic characteristic data of participants involved in this study are summarized in Table 1. Mean age and BMI were not significantly different between non-diabetic and diabetic participants. Diabetic participants displayed elevated HbA1c and FBS and lower C-peptide levels.
| Biochemical Parameters |
Non-diabetic Participants Mean ± SE |
Diabetic Participants Mean ± SE |
df | t | P-value |
|---|---|---|---|---|---|
| Glucose (mmol/L) | 5.38±0.14 | 8.45±0.41 | 52.88 | 7.10 | 0.001* |
| HbA1c (%) | 5.40±0.78 | 7.95±0.85 | 98 | 4.57 | 0.001* |
| C-peptide (ng/mL) | 2.02±0.02 | 1.96±0.01 | 83.21 | -1.92 | 0.05* |
| GOI |
Non-diabetic Participants Mean ± SE |
Diabetic Participants Mean ± SE |
df | t | P-value |
|---|---|---|---|---|---|
| GSK3 | 0.49±0.03 | 0.46±0.02 | 98 | -0.722 | 0.472 |
| - | Sum of Squares | df | Mean Square | F | Sig. | |
|---|---|---|---|---|---|---|
| BMI Categories | Between Groups | 0.026 | 3 | 0.009 | 0.202 | 0.895 |
| Within Groups | 4.142 | 96 | 0.043 | - | - | |
| Total | 4.169 | 99 | - | - | - | |
| Study Variables | Non-diabetic Participants | Diabetic Participants | |||
|---|---|---|---|---|---|
| Pearson Correlation (r) | P-value | Pearson Correlation (r) | P-value | ||
| C-Peptide | -0.045 | 0.762 | -0.204 | 0.155 | |
| FBS | -0.034 | 0.817 | 0.358* | 0.012 | |
| HbA1c | -0.122 | 0.408 | 0.181 | 0.210 | |
| Gender | 0.159 | 0.285 | 0.151 | 0.299 | |
| Age | -0.208 | 0.161 | -0.170 | 0.249 | |
| BMI | 0.035 | 0.813 | 0.047 | 0.747 | |
| Family History | -0.139 | 0.357 | 0.071 | 0.640 | |
Gene expression level of GSK3 was higher in Type II diabetic participants compared to the non-diabetics, but it was not statistically significant (Table 2).
Each diabetic and non-diabetic group of participants was further divided into two groups based on the BMI (BMI<30 and BMI>30) to find out the possible role of obesity on the regulation of GSK3 expression. Of the diabetics, 50% were normal/overweight (BMI <30 kg/m2) and 50% were obese (BMI of >30 kg/m2). In non-diabetic participants, 50% were normal/overweight and 50% were obese. This weight distribution was not significantly different between the four groups. To compare differences between GSK3 expression levels based on BMI categories in diabetic and non-diabetic participants, one-way ANOVA was used in which no significant difference was found (Table 3).
Diabetic and non-diabetic participants were divided into four groups based on family history of diabetes, and it was found that 25 (50%) non-diabetic participants and 29 (58%) diabetic participants had a family history of Type II diabetes. Pearson's correlation analysis was performed to investigate the plausible relationship between GSK3 expression and family history of Type II diabetes. No significant correlation was found in the gene expression of GSK3 based on a family history of diabetes in diabetic and non-diabetic participants (Table 4).
To investigate the relationships between GSK3 gene expression and indicators of insulin resistance, Pearson's correlation analysis was performed, which indicated no significant association between GSK3 expression and indicators of insulin resistance in two groups in except a significant positive correlation between GSK3 expression and FBS in diabetic participants (Table 4).
4. DISCUSSION
GSK3 is a serine/threonine kinase that is involved in the storage of glucose into glycogen. Defects in GSK3 and GS function are early stages of the development of insulin resistance, which may cause impaired glycogen synthesis in T2DM [26].
GSK3 activity has been shown to be increased in skeletal muscle and adipose tissue of insulin-resistant animals [26, 27]. In obese Zucker rats, reduced phosphorylation of GSK3 has been reported [27]. Constitutive activation of GSK3 in vitro [28] or muscle-specific overexpression of GSK3 [25] resulted in insulin resistance. Inhibition of GSK3 in Zucker diabetic fatty rats leads to improvement in insulin function and glucose uptake [7]. It has been demonstrated that PI3K/AKT pathway plays a key role in insulin-stimulated glucose transport [26, 29-31]. Reduced insulin stimulated glucose transport is one of the major metabolic defects of skeletal muscles in insulin-resistant and type II diabetic individuals [29-31]. Previous studies have demonstrated that the overexpression of GSK3 in skeletal muscle samples from insulin resistance states may negatively influence insulin signal transduction and cause its weakness, particularly at IR and IRS [32]. Also, overexpression of GSK3 in either cultured cells [28] or mice [28] has been shown to antagonize insulin signaling. One mechanism whereby GSK3 increases insulin resistance is by serine phosphorylation of IRS, resulting in reduced tyrosine phosphorylation of IRS by the insulin receptor and reduced PI3K/AKT signaling to downstream components [33]. Others have found that insulin resistance may be contributed to defects in glycogen synthesis [34]. GSK3 gene expression levels and function are increased in muscles of Type II diabetics patients and are inversely correlated with both GS function and insulin-stimulated glucose transport [10, 14]. Treatment of both human muscle cells and insulin-resistant rat with GSK3 inhibitors can increase insulin-stimulated glycogen synthesis and glucose transport [35, 36]. Transgenic mice with muscle-specific overexpression of human GSK3 developed glucose intolerance and hyperlipidemia [25].
In contrast to these findings, our study demonstrated no significant difference in GSK3 gene expression between diabetic and non-diabetic participants. Additionally, there was no significant difference in GSK3 gene expression based on BMI categories and family history of diabetes. Although correlations do not prove causal relationships, there was an expected positive correlation between GSK3 gene expression level and FBS only in diabetic participants. No association between GSK3 gene expression levels and indicators of insulin resistance was observed in non-diabetic participants. One possible interpretation of this observation is that higher gene expression levels of GSK3 in diabetics (although it was not enough to reach a significant amount) may predispose them to failure in glucose utilization and the development of insulin resistance. To understand more about the involvement of GSK3 in obesity and insulin resistance, the correlation between GSK3 gene expression level and BMI level was considered and no significant correlation was found. These results indicate that regulation of GSK3 may occur at the level of translation, as gene expression levels were unaltered between diabetic and non-diabetic as well as based on BMI categories or family history of diabetes. Whether disturbances in GSK3 expression or function are a cause of insulin resistance or a consequence of that, is unknown.
This study is consistent with previous studies which found no changes in GSK3 activity in samples from diabetic human or animals [20, 35, 37] and differ from those that found elevated GSK3 activity [38-40]. Information about the regulation of GSK3 in humans is limited. Acute bouts of exercise have been shown to both increase [15] and decrease [41] GSK3 activity in muscle. Differences in the duration and intensity of the exercise may be important variables. Prolonged treatment of obese individuals with impaired glucose tolerance with diet and exercise program caused reduced GSK3 gene expression in skeletal muscles [42].
Contradictions in literature could be due to the differences in experimental design, the method of tissue collection, and processing, which requires considerable optimization to overcome. Since this study merely considered the gene expression regulation without post-translation modification and tissue-specific differences, it is difficult to draw conclusions on the role of defective GSK3 function in the development of insulin resistance.
Our data indicate that it is probable that glycogen synthase activity is regulated by non-insulin mediated mechanisms such as exercise [43] or allosteric changes independent of GSK3 expression [44].
Since many functions are regulated by GSK3, the activity of GSK3 must be highly regulated. The main mechanisms of regulating the actions of GSK3 in a substrate-specific manner are [45]: regulation by GSK3 autophosphorylation, phosphorylation state of GSK3 substrates, translocation of GSK3, and formation of protein complexes containing GSK3 that direct, or inhibit, its actions toward specific substrates. The most well-defined regulatory mechanism is inhibition of the activity of GSK3 by phosphorylation of a regulatory serine [46]. The PI3K/AKT signaling pathway, activated in response to insulin, is one of the main regulators of GSK3. AKT and protein kinase C phosphorylates/inhibits GSK3 on serine residues [9]. Conversely, the enzymatic activity of GSK3 is stimulated by tyrosine-phosphorylation, but the mechanisms underlying this modification are not well-defined. This autoposhorylation of the tyrosine residue is probably intramolecularly mediated and not under the regulatory control of upstream modulators [9].
Since GSK3 is constitutively stimulated and activated [8], its regulation might be achieved mainly through the inhibitory serine-phosphorylation. Although normal suppression of GSK3 is controlled by insulin, the overstimulation of receptors may cause insensitivity of the GSK3 and result in uninhibited GSK3 activity [11]. Therefore, the release of GSK3 activity, a hallmark of PI3K/AKT/GSK3 pathway insensitivity, can happen under disease conditions as a consequence of overstimulation or inhibition of PI3K/AKT. These diversity mechanisms controlling the actions of GSK3 provide substrate-specific control of its functions, and exclusive capacity for an enzyme that can regulate numerous cellular functions.
Moreover, it has been found that the circulating levels of both glucose and insulin regulate GSK3 activity. This complicated relationship and collaboration between insulin and glucose may cause contradictions in literature as well. Also, as insulin depletion and hyperglycemia lead to tissue-specific changes, the activity of GSK3 might vary among different tissues [32, 47]. High fat diet induced-diabetes in mice caused a slight reduction in the activity of GSK3 in the liver, enhancement in epididymal fat, and no changes in skeletal muscle [38], indicating that regulation of GSK3 in insulin resistance conditions might be different among tissues [32, 48]. Also, it has been revealed that changes in GSK3 phosphorylation due to insulin deficiency in diabetic mice brain are big and opposite to changes in peripheral tissues as phosphorylation level of GSK3 in the brain is increased while reduced in epididymal fat [49]. This enhanced GSK3 phosphorylation is ascribed to hyperglycemia associated with insulin depletion. Hyperglycemia, in the presence of normal or deficient insulin levels, caused increased GSK3 phosphorylation level, whereas it was reduced by hypoglycemia with insulin administration [49]. Thus, both insulin and glucose contribute to regulating the activity of GSK3.
Also, it has been demonstrated that using GSK3 knock-in mice, in which AKT phosphorylation sites on GSK3α and GSK3β were mutated, did not affect blood glucose level and insulin-induced hepatic glycogen accumulation were normal [50]. Also, a regulatory axis consisting of AKT/PPP1R3G/ PPP1R3B that controls glycogen synthesis and glucose homeostasis in parallel to the GSK3-dependent axis has been revealed [51]. In that study, the knocking down of PPP1R3G inhibited insulin response while the wild type PPP1R3G, and not phosphorylation-defective mutants, increased glycogen deposition and insulin sensitivity. PPP1R3G, as a regulatory subunit of protein phosphatase1 (PP1), is phosphorylated by AKT, where its phosphorylation fluctuates with the fasting-feeding cycle and is necessary for insulin-stimulated glycogen synthase. Consequently, a GSK3-independent pathway has been proposed to link insulin signaling with glycogen synthesis.
CONCLUSION
Even though several lines of evidence support the involvement of GSK3 in insulin resistance, the role of GSK3 in diabetes is currently unknown and requires further investigations.
Although increased serine phosphorylation of GSK3 represents a mechanism for the control of GSK3 activity in response to hormones, such as insulin, it is probable that glycogen synthase activity is regulated by non-insulin mediated mechanisms as well, such as exercise or allosteric changes independent of GSK3 expression or other insulin-dependent pathways. Taken together, it appears that there is tissue specificity for the expression and post-translation regulations of GSK3, which may cause hyperactivation or overexpression in some target tissues in diabetes, since circulating levels of both glucose and insulin regulate GSK3 activity.
AUTHORS' CONTRIBUTIONS
SAHK1, MSAM1, and HK6 were responsible for the study concept and design. SAHK1 and JAA3 contributed to data acquisition. MFS2 assisted SAHK1 with data analysis and interpretation of findings. SAHK1 drafted the manuscript. MSAM1, HK6, ZR4, and RMA5 provided critical revision of the manuscript for important intellectual content and approved the final version for publication.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The necessary approval was obtained from the University Putra Malaysia Ethics Committee, Malaysia, Ref. no: UPM/TNCPI/RMC/JKEUPM/1.4.18.1/F1.
HUMAN AND ANIMAL RIGHTS
No animals were used in this study. The study on humans was conducted in accordance with the Declaration of Helsinki in its currently applicable version, the guidelines of the International Conference on Harmonization of Good Clinical Practice (ICH-GCP) and coordination with the Health Ministry of Malaysia was fulfilled based on the applicable Malaysian laws (National Medical Research Register, (NMRR)).
CONSENT FOR PUBLICATION
Consent has been obtained from each participant after a full explanation of the purpose and nature of all procedures used.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available within the study.
FUNDING
This work was supported by the Fundamental Research Grant Scheme (grant number: 14-554-20427).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.
